Die Thalassämie ist eine Erkrankung, welche von den Eltern auf das Kind übertragen wird. Nach Angaben der Weltgesundheitsorganisation sind „die Alpha und die Beta-Thalassämie, die am häufigsten vererbten Einzel-Gen Erkrankungen auf der Welt.“
Laut Studien der Universität von Harvard müssen beide Elternteile ein Gen für Thalassämie tragen, damit das Kind auf eine Transfusion angewiesen sein wird. Demnach, besteht ein Risiko von 25%, dass das Kind an eine schwere Anämie, die sogenannte „Thalassämie major“, erkranken wird. Zur Veranschaulichung der genetischen Verteilung siehe das folgende Diagramm:
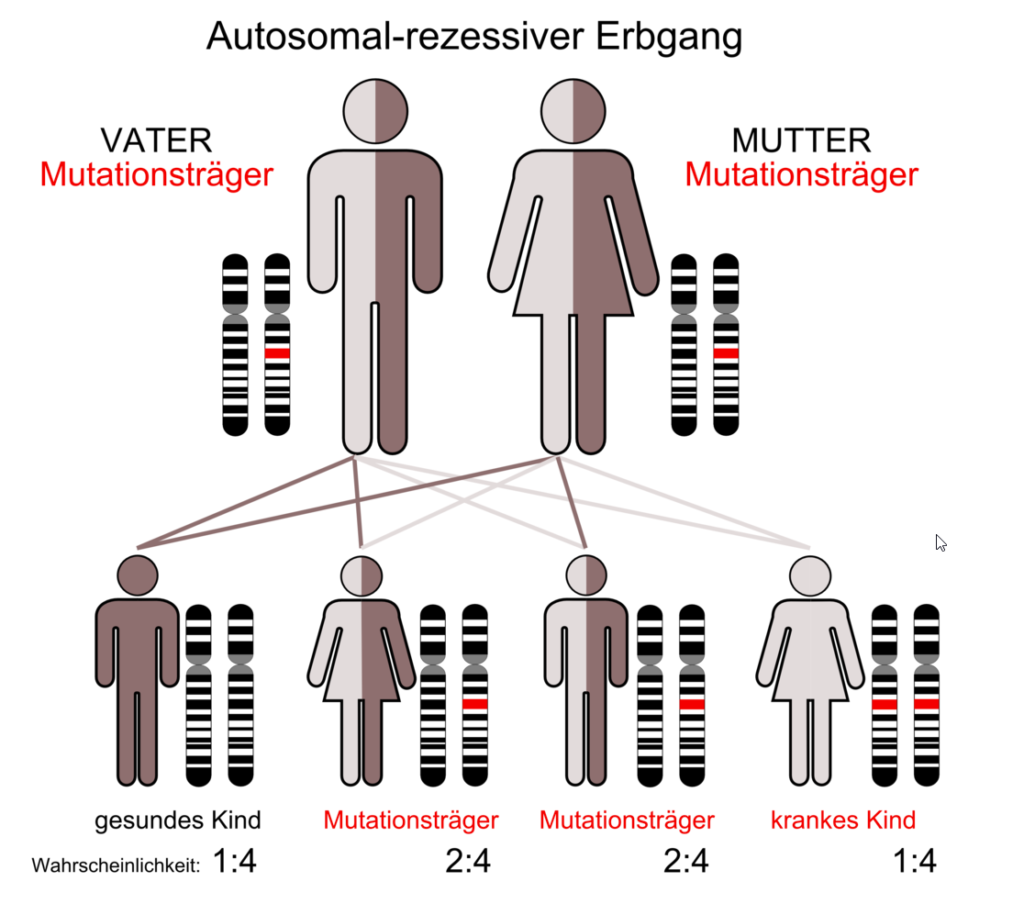
ie Sie der Grafik entnehmen können, kann ein Kind mit Thalassämie an unterschiedlichen Variationen oder Schweregrade der Erkrankung leiden. Wie bereits erwähnt, ist die Thalassämie major der schwerste Typ und gilt als seltene Erkrankung, da nur ein sehr geringer Prozentsatz der Bevölkerung darunter leidet.
Das Knochenmark eines Kindes, bei dem Thalassämie major diagnostiziert wurde, produziert nicht ausreichend gebildete rote Blutkörperchen, das sogenannte Hämoglobin, und dies erschwert ihm ein selbstständiges Leben. Um leben und wachsen zu können, sind die betroffenen Kinder jede 2-4 Wochen auf lebenslange Transfusionen angewiesen.
Jedes rote Blutkörperchen enthält ein Eisenmolekül. Wenn eine Person chronische Bluttransfusionen erhält, lagern sich diese Eisenmoleküle im Köper ab. Der einzige Weg das Eisen aus dem Körper zu entfernen, ist mittels Medikamente. Es gibt verschiedene Sorten von Medikamente, die eine Eisen-Ausscheidung aus dem Körper ermöglichen (in Tabletten oder in flüssiger Form).
Die medizinische Forschung hat in den letzten 20 Jahren zahlreiche Errungenschaften ermöglicht. Wir können uns glücklich schätzen, dass wir in einer Zeit leben, in der bahnbrechende und vielversprechende Therapien erforscht werden. Gegenwärtig jedoch, ist Thalassämie nur durch eine Knochenmarktransplantation eines nahezu perfekt übereinstimmenden Geschwisterspender heilbar.
Mit der richtigen Transfusionen und Chelat-Einhaltung kann ein Kind mit Thalassämie zu einem voll funktionsfähigen Erwachsenen werden. Dank der heutigen fortgeschrittenen Medizintechnik sind keine Grenzen gesetzt, für jemanden, der mit Thalassämie geboren wurde.