Thalassemia is a disease that is transmited from parent to child. According to the World Health Organization, “alpha and beta thalassemia are the most commonly inherited single-gene disorders in the world.”
4
According to studies by Harvard University, both parents must carry a gene for thalassemia for the child to need a transfusion. Accordingly, there is a 25% risk that the child will develop severe anemia, known as “thalassemia major”. To illustrate the genetic distribution, see the following diagram:
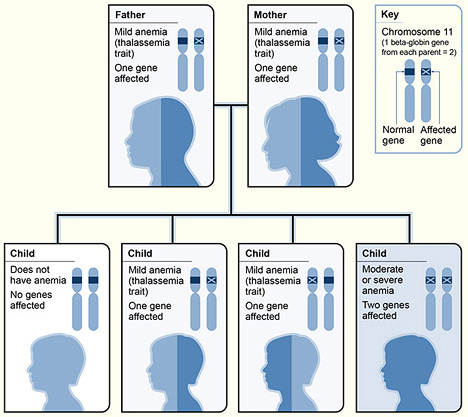
As you can see from the graph, a child with thalassemia can suffer from different variations or severities of the disease. As mentioned above, thalassemia major is the most severe type and is considered a rare disease as only a very small percentage of the population suffers from it.
The bone marrow of a child diagnosed with thalassemia major does not produce enough red blood cells, known as hemoglobin, and this makes it difficult for the child to live independently. In order to live and grow, the affected children are dependent on lifelong transfusions every 2-4 weeks.
Each red blood cell contains an iron molecule. When a person receives chronic blood transfusions, these iron molecules accumulate in the body. The only way to remove the iron from the body is through medication. There are different types of drugs that allow iron excretion from the body (in tablet or liquid form).
Medical research has made numerous achievements possible in the last 20 years. We are fortunate to live in a time when groundbreaking and promising therapies are being researched. Currently, however, thalassemia can only be cured by a bone marrow transplant from an almost perfectly matched sibling donor.
5
With the right transfusions and chelation compliance, a child with thalassemia can become a fully functioning adult. Thanks to today’s advanced medical technology, there are no limits for someone born with thalassemia.